

Sickle cell disease (SCD) is a blood disorder where the body produces abnormally shaped red blood cells (RBCs).
SCD is associated with severe pain and complications1 that can affect the entire body.
SCD Handbooks/Guides for caregivers:
Normal red blood cells (RBCs) are round, smooth, and flat like a plate. They flow through blood vessels very easily. RBCs that carry sickle hemoglobin:
Problems from blocked blood flow can happen quickly, such as sudden pain. Other problems happen more slowly and can damage the body’s tissues and organs over time.
Another problem with sickle cells is that they do not live as long as normal RBCs.
There are several different types4 of sickle cell disease (SCD) with varying symptoms, complications, and severities.
The 4 most common types of SCD are:
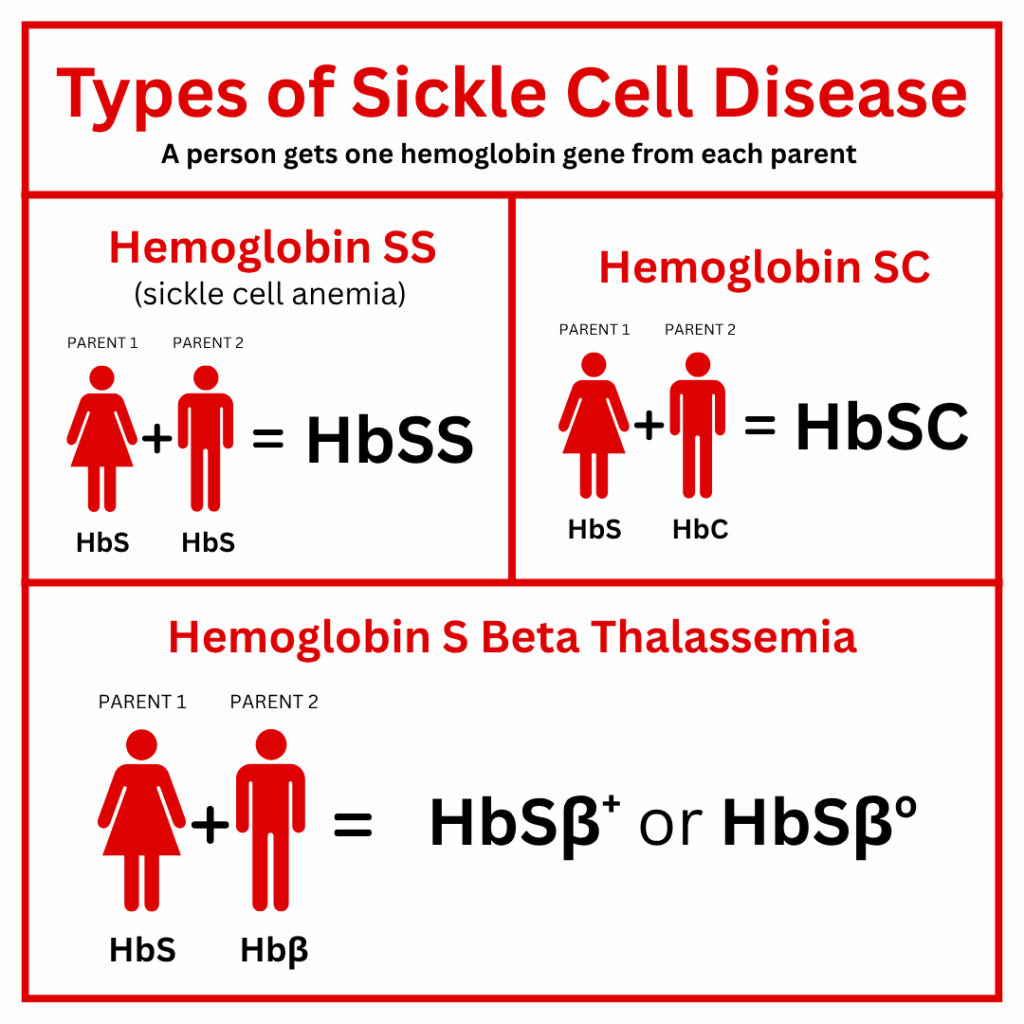
Beta (β) Thalassemia is another type of gene a person receives from a parent who has the Thalassemia trait (Aβ).
Patients and families should watch for the following conditions that need an urgent medical evaluation:
• Fever of 101° F or higher
• Chest pain
• Shortness of breath
• Increasing tiredness
• Abdominal swelling
• Unusual headache
• Any sudden weakness or loss of feeling
• Pain that will not go away with home treatment
• Priapism (painful erection that will not go down)
• Sudden vision change
Sickle cell disease (SCD) is a blood disorder where the body produces abnormally shaped red blood cells (RBCs).
SCD is associated with severe pain and complications1 that can affect the entire body.
SCD Handbooks/Guides for caregivers:
Normal red blood cells (RBCs) are round, smooth, and flat like a plate. They flow through blood vessels very easily. RBCs that carry sickle hemoglobin:
Problems from blocked blood flow can happen quickly, such as sudden pain. Other problems happen more slowly and can damage the body’s tissues and organs over time.
Another problem with sickle cells is that they do not live as long as normal RBCs.
There are several different types4 of sickle cell disease (SCD) with varying symptoms, complications, and severities.
The 4 most common types of SCD are:
Beta (β) Thalassemia is another type of gene a person receives from a parent who has the Thalassemia trait (Aβ).
Patients and families should watch for the following conditions that need an urgent medical evaluation:
• Fever of 101° F or higher
• Chest pain
• Shortness of breath
• Increasing tiredness
• Abdominal swelling
• Unusual headache
• Any sudden weakness or loss of feeling
• Pain that will not go away with home treatment
• Priapism (painful erection that will not go down)
• Sudden vision change
Sickle cell disease (SCD) is a blood disorder where the body produces abnormally shaped red blood cells (RBCs).
SCD is associated with severe pain and complications1 that can affect the entire body.
SCD Handbooks/Guides for caregivers:
Normal red blood cells (RBCs) are round, smooth, and flat like a plate. They flow through blood vessels very easily. RBCs that carry sickle hemoglobin:
Problems from blocked blood flow can happen quickly, such as sudden pain. Other problems happen more slowly and can damage the body’s tissues and organs over time.
Another problem with sickle cells is that they do not live as long as normal RBCs.
There are several different types4 of sickle cell disease (SCD) with varying symptoms, complications, and severities.
The 4 most common types of SCD are:
Beta (β) Thalassemia is another type of gene a person receives from a parent who has the Thalassemia trait (Aβ).
Patients and families should watch for the following conditions that need an urgent medical evaluation:
• Fever of 101° F or higher
• Chest pain
• Shortness of breath
• Increasing tiredness
• Abdominal swelling
• Unusual headache
• Any sudden weakness or loss of feeling
• Pain that will not go away with home treatment
• Priapism (painful erection that will not go down)
• Sudden vision change